Western blotting or immunoblotting is an indispensable technique, almost every published paper in area of molecular cell biology uses western blotting for detecting specific proteins in a sample of tissue homogenate or cell lysates. Western blotting combines resolving power of polyacrylamide gel electrophoresis (PAGE) or SDS-PAGE and specificity of antibodies to detect target proteins. Proteins are resolved on the basis of their molecular weight in SDS-PAGE and transferred from the polyacrylamide gel onto the membranes (Nitrocellulose or PVDF), which creates an exact replica of the protein separation pattern on the membrane. After transferring the proteins to the membrane, the membrane needs ‘blocking’ to ‘block’ non-specific binding sites on the surface of the membrane. Blocking is usually performed with Bovine Serum Albumin, Skimmed milk or purified milk Casein.
G-Biosciences recommend any of the following blocking agents for blocking the membranes.
In absence of blocking, antibodies will bind to nonspecific binding sites on the membrane and will make it difficult to detect the presence of the target protein, leading to false positives. Once proteins have been immobilised on the binding membrane after blocking, they can be probed with a primary antibody, which is specific to the protein of interest. Adsorbed or bound proteins can be detected either with a primary antibody coupled to a reporter molecule or a secondary antibody coupled to the a reporter molecule such as Horseradish peroxidase or a fluorophore.
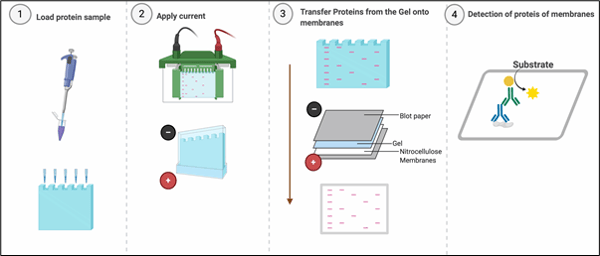
How to Prepare Your Samples ?
When preparing samples for Western blot analysis, following instructions need to be adhered to
- Choose the Right Lysis Buffer for Your Sample
To ensure the success of the western blot experiment, one must choose the correct lysis buffer, the choice of lysis buffer should be made based on the requirements of the experiment. This is because lysis buffer composition has a profound impact on solubilisation and extraction of proteins from different subcellular compartments. For example RIPA buffer performs well for extraction of cytoplasmic proteins but is not a good choice for extraction and solubilisation of cytoskeletal and extracellular matrix proteins.
Choosing the proper lysis buffer depends on the subcellular localization of the protein. The process of selecting a lysis buffer and determining an appropriate volume is usually a process of trial and error and it is highly recommended to confirm proper solubilisation of target protein by trying a couple of lysis buffers before using lysates for the experiment.
- Whether to add detergents or not ?
Adding detergents to the lysis buffer can solubilise proteins of interest. Lysis buffers can be supplemented with non-ionic or ionic detergents or both to solubilise and extract the protein of interest. Again the lysis buffers should be supplemented with different detergents to optimise cell lysis buffer for a right choice.
- Add Protease and Phosphatase Inhibitors
Cell or tissue lysis release enzymes that are otherwise sequestered in different subcellular compartments and doesn’t interact with each other under natural conditions. For example, cell lysis release proteolytic enzymes which would digest proteins and therefore it is important to add protease inhibitors. Similarly it is also important to add phosphatase inhibitors to preserve native phosphorylation status of the proteins. Protease and phosphatase inhibitors prevent fast proteolysis and dephosphorylation of the target proteins upon cell lysis. One must prepare fresh lysis buffers before every experiment and supplement them with Protease and phosphatase inhibitors from stock. During the lysis cells and buffers should be at 4°C during the entire procedure.
- Determine Protein Concentration before you load your samples
To ensure equal loading of cellular lysate in SDS polyacrylamide gels, one must determine the protein concentration of each lysate and normalize it across the samples of every experiment. With equal loading, one can accurately quantify protein levels and expression differences by Western blotting. To determine protein concentration, you can use a variety of protein assay methods, such as the Lowry assay, the Bradford assay, the BCA assay or even UV spectroscopy. We advise loading approximately 20-30 μg of the protein lysate to ensure a linear response between band intensity and amount of protein loaded which tends to become hyperbolic when protein lysate is loaded in excess.
- Load the Proteins
Add an equal volume of SDS-PAGE Sample Loading Buffer [2X] to the tube containing protein solution. For reducing gels, add reducing agent to a final concentration of 2-5% β-mercaptoethanol or 5-20mM DTT. Vortex the tube to mix the contents. Heat the sample tubes for 5-10 minutes at 95°C. After the boiling is complete, vortex and centrifuge the tube for 30 seconds. The sample is now ready for loading on SDS-PAGE gels. Vortex the tube before loading the protein solution on the gel.
Once protein concentration has been determined, one must dilute the protein samples in the gel loading buffer, otherwise known as 2x Laemmli sample buffer which has following composition
65.8 mM Tris-HCl, pH 6.8
26.3% (w/v) glycerol
2.1% SDS (w/w)
0.01% bromophenol blue (w/v)
Glycerol helps samples sink easily into the wells of the gel. The buffer contains bromophenol blue, a tracking dye that reaches the bottom of the gel that indicates the end of electrophoresis. SDS is a denaturant which binds to the proteins and imparts a negative charge. SDS binding allows denaturation of proteins and allows migration of proteins according to molecular weight. Before loading the samples, you must heat them for 5 minutes at 100°C, or 10 minutes at 70°C to aid in the denaturation. Once the samples are heated, they can be loaded into the gels immediately, or placed at 4°C or -20°C for later analysis.