A proteome is a sum total of all the cellular proteins, this would include housekeeping proteins and proteins that are produced in response to a stimulus. Some proteins are produced only in a given cell type or tissue, others are produced in all cells, some proteins are only produced under certain given conditions adding to the complexity and heterogeneity of proteins in a proteome. Similarly, alternative splicing and post translational modifications all together combined can make the proteome very complex.
The abundance of proteins in a biological sample can vary greatly from just 100 proteins to 106 proteins in a humble Saccharomyces cerevisiae cell. Another factor is the dynamic range of proteins, the proteins can vary in their relative abundance in a proteome to great extent, implies that proteins can have anywhere from just 1 copy to ten million copies of a given protein. Thus proteins tend to have a very wide dynamic range ranging from 5 to 12 orders of magnitude. Modern mass spectroscopic devices can cover a dynamic range of only 2-3 orders and consequently a large number of proteins are destined to go undetected.
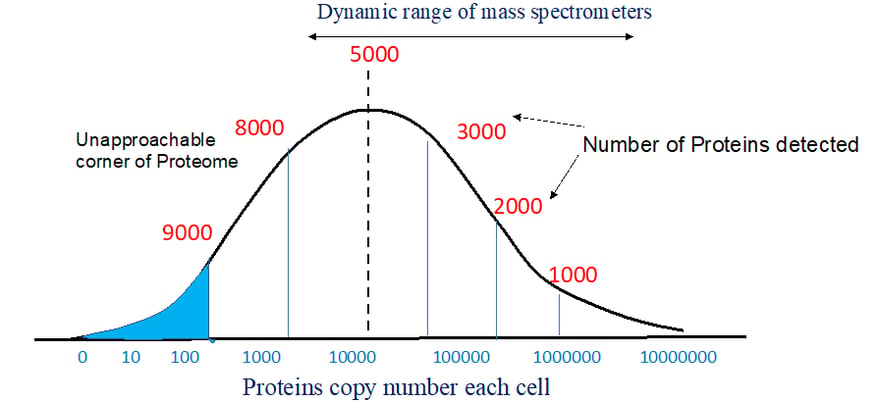
Figure 1. Distribution of proteins in sample and dynamic range.
Therefore without appropriate sample preparation strategies that aim at reduction of dynamic range and complexity of proteome sample, analysis of proteins samples cannot be done to retrieve most relevant information. Specially important is the problem of low abundance proteins, the low abundance proteins are likely to remain undetected because the distribution of proteome abundance is nearly a bell shaped distribution curve on a logarithmic copy number scale. Because of this symmetric distribution trend, the mid-point of bell coincides with the maximum of the distribution dividing into two equal halves. Figure 1 shows that exploring the most abundant half of the proteome leads to detection of an increasing number of proteins for every order of magnitude increase in sensitivity or sample size but once this region has been detected i.e the maximum half has been detected, the slope of the distribution inverts, and for every order of magnitude increase yields a progressively smaller number of proteins are detected. The real challenge is the identification of proteins sitting at the “corner” of the proteome, which is made up of more than 1000 proteins of extremely small abundance (See Figure 1).

Figure 2. Reducing proteome complexity by removal of high abundance proteins. Exploiting mass distribution and enzyme kinetics of trypsinization to prepare a sample to capture low abundance proteins.
Therefore preparation of protein samples is tricky and no single technique or method works for all applications. One has to rationally understand the biophysical and biochemical properties of the proteins to enable oneself to apply methods that exploit parameters such as size, shape, charge, solubility, stability, sedimentation velocity, affinity for certain substrates to fractionate proteins of interest from the complex proteome samples. Moreover one may require to use more than one techniques or methods to achieved the same, very often the complex samples can be ‘reduced’ to simple samples by using many protocols used in end to end manner. One such approach is summarized in figure 2. The figure 2 shows that proteins are digested by trypsin in a specific manner using very high dilutions, which ensures that trypsin primarily works on the high abundance proteins. Under this kind of arrangement the high-abundance proteins are preferentially digested at first according to Michaelis-Menten kinetics, which are then removed by molecular weight cut-off spin filters. The overall outcome is removal of high abundance proteins from the proteome sample, which then leads to reduction in complexity of sample and enables identification of low abundance proteins by mass spectroscopes.
References:
- Zubarev RA. The challenge of the proteome dynamic range and its implications for in-depth proteomics. Proteomics. 2013 Mar;13(5):723-6.
- Picotti P, Bodenmiller B, Mueller LN, Domon B, Aebersold R. Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell. 2009 Aug 21;138(4):795-806 3.
- Fonslow BR, Stein BD, Webb KJ, Xu T, Choi J, Park SK, Yates JR 3rd. Digestion and depletion of abundant proteins improves proteomic coverage. Nat Methods. 2013 Jan;10(1):54-6.