This is a follow up blog to SILAC for Improved Mass Spectrometry & Quantitative Proteomics
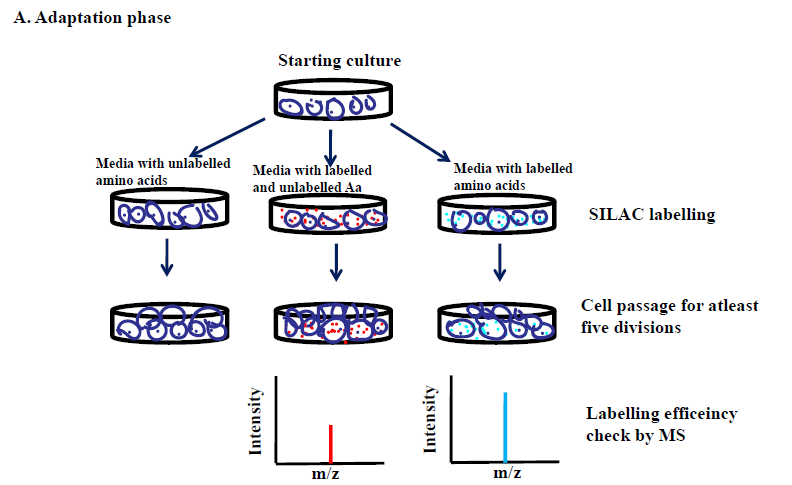
SILAC (Stable Isotope Labeling with Amino Acids in Cell Culture) method of working is differentiated into two critical phases: an adaptation phase and an experimental phase. Before initiating the adaptation phase, it is crucial to characterize the cell type to be used for the labeling. In general, dialyzed serum is used to rear the cells in order to negate the availability of free amino acids present in the normal serum. Although, some cell lines do not grow that well in a dialyzed medium due to the absence of some of the growth factors, therefore, supplementation of purified growth factors with the dialyzed media or small percentage of normal serum in the dialyzed media may be helpful.
Adaptation phase
The selected cells are acclimatized in the unlabeled and labelled media until all the cellular proteins in the labelled group are maximally incorporated with the heavy amino acids (95% or more). The cells are then allowed to divide for at least five cell divisions. A small proportion of the heavy labelled cell population is harvested and the cells are digested into small peptides using trypsin followed by evaluation of incorporation of heavy amino acids by LC-MS/MS.
If the efficiency is not high enough, the cells are allowed to grow further and rechecked for efficiency evaluation until the desired labeling is achieved.
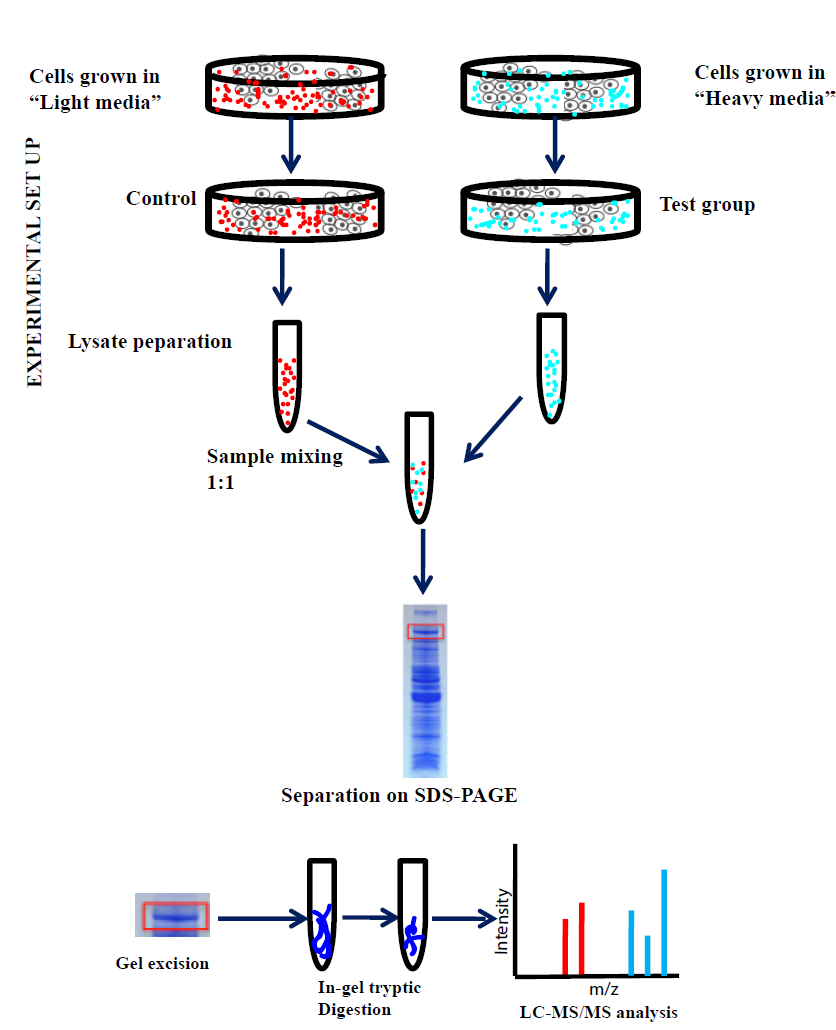
Experimental phase
Once the incorporation of heavy amino acids is ensured, the two cell populations can be subjected to tests according to the experimental plans. The cell populations are then combined in equal proportions before subjecting to optional methods, such as subcellular organelle purification, cell lysis, protein extraction, and protein digestion. The fragmented samples are further analysed with LC-MS/MS and the quantification is done by comparing the ratios of heavy peptides to light peptides. Generally to perform the SILAC-MS analysis, Orbitrap-based mass spectrometers, such as linear ion-trap Orbitrap (LTQ-OrbitrapVelos) or quadrupole Orbitrap (Q-Exactive), which gives an advantage of higher resolving power, sequencing speed, dynamic ranges and mass accuracy. Different proteome discovery software such as MaxQuant, Census, Trans-proteomic pipeline (TPP), and pQuant can be used to delineate the high-quality MS data and quantification of the peptides. After all the quantification, in order to develop insight into the proteomic results obtained from MS data, annotation databases such as GO, KEGG, STRING, or bioinformatics tools such as GoMiner, cytoscape, DAVID can be applied.
Why to choose SILAC?
 SILAC shares greater advantages in proteome quantification over other labelling methods especially for extensive sample treatment and processing, such as affinity purification of protein complexes, subcellular fractionation or posttranslational modifications. Moreover, SILAC can be used to analyse a wide varieties of samples, such as cells, tissues, and body fluids (spike-in SILAC and super-SILAC). It can be implemented easily and is robust in terms of data analysis and result interpretation. Over other labeling methods, SILAC has more proteome quantitative reliability.
SILAC shares greater advantages in proteome quantification over other labelling methods especially for extensive sample treatment and processing, such as affinity purification of protein complexes, subcellular fractionation or posttranslational modifications. Moreover, SILAC can be used to analyse a wide varieties of samples, such as cells, tissues, and body fluids (spike-in SILAC and super-SILAC). It can be implemented easily and is robust in terms of data analysis and result interpretation. Over other labeling methods, SILAC has more proteome quantitative reliability.
SILAC has only few disadvantages of SILAC as it offers a limited number of cellular states for comparison contrary to iTRAQ or TMT labelling. However, this problem can be dealt by clubbing several SILAC experiments in same testing approach, which can allow an analysis for almost nine-point dynamic signalling pathways.
In-Gel Tryptic Digestion for SILAC peptide preparation:
In order to make peptide fragments for proteome quantification, it is essential to develop a suitable digestion protocol for precise identification and quantification of the proteome. In general, an enzymatic protein or a chemical can be used for site specific cleavage of the amino acid sequence into fragments of peptides of varied lengths. The exact mass of the fragments can be quantified by MS/LC-MS to allow a robust and precise proteome analysis. Though, a wide range of proteases and chemicals are available commercially, Trypsin is the most commonly used proteolytic enzyme for digestion. It is a serine protease specific for the digestion of C-terminus side of arginine or lysine, subsequently producing tryptic peptides of varied lengths. The location of these basic amino acids (arginine and lysine) at the C-terminus of the tryptic peptides enhances ionization of the fragments leading to an enhanced signal intensity of the mass spectral peaks in MS/LC-MS.
Tryptic digestion can be performed in-solution or in-gel depending on the sample availability and its complexity. For small sample volumes with lesser complexity, In-solution digestion is preferred. While for very complex samples, in-gel digestion is preferred as it combines both digestion and separation in same workflow. It provides an enhanced visual indication of protein availability relatively. Both in-solution and in-gel sample processing kits are commercially available.
In-gel Tryptic digestion method
Large scale In-gel tryptic requires initial denaturation and separation of the protein samples on a 1D-polyacrylamide denaturation gel using commercially available kits or in house reagents. The gel subsequently can be excised into small squares (2x2 mm) and submerged into a destaining solution (0.2% ammonium bicarbonate in 50% acetonitrile), subsequently incubated at 37°C for 30 minutes with constant shaking. The destaining solution must be changed after that until the gel slices gets completely destained. The destained gel pieces are then added with a reducing buffer (200 μl of TCEP in 2 ml Digestion buffer) and heated at 60°C for 10 minutes. The slices are then cooled at RT and the reducing buffer is discarded after that. An alkylation buffer (60 mg of iodoacetamide in 3 ml Digestion buffer) is further mixed with the gel pieces and incubate in dark for 1 hour at room temperature. Once the alkylation is completed, the solution is washed twice with the digestion buffer for 15 minutes at 37°C in order to remove excess alkylation buffer. The gel pieces are then shrunk using acetonitrile for 15 minutes at room temperature followed by air drying of the gel pieces at room temperature for 15 minutes. The gel pieces are then swollen by using of Activated Trypsin solution at room temperature for 15 minutes. The trypsin solution after proteolytic digestion is diluted with Digestion Buffer and incubated at 37°C for 4 hours or at 30°C overnight with constant stirring. The digestion buffer is taken out post incubation and stored in a fresh tube for further use. In order to extract the digested peptides, 5% formic acid solution is added to the gel pieces for 5 minutes. Another step of acetylation is carried out after removal of digestion buffer. The acetonitrile solution is washed with the saved digestion mixture. For preparation of peptides for MS analysis, the tryptic digested peptides are lyophilised and reconstituted in 0.1% formic acid. It should be noted that for the entire process all reagents should be freshly prepared and clean tubes should be used.
|
Quantification method |
Sample processing |
Labelling |
Level of comparisons |
Accuracy level |
Proteome coverage |
Range of analysis |
|
SILAC (metabolic labelling) |
Ex-vivo or in-vivo (Cell cultures, tissues or animal models) |
Protein samples |
Up to 5 (can be used in combinations) |
very high |
high |
1-2 log |
|
15N (metabolic labelling) |
Cells |
Protein samples |
2 |
high |
high |
1-2 log |
|
ICAT (chemical labelling) |
in-vitro (cells, animal tissues) |
Protein samples |
2 |
high |
high |
2 log |
|
iTRAQ (chemical labelling) |
in-vitro |
peptides |
up to 4-8 |
high |
high |
2 log |
|
TMT (chemical labelling) |
in-vitro |
peptides |
up to 6-10 |
high |
high |
2 log |
|
Dimethyl labelling (chemical labelling) |
in-vitro |
peptides |
2-3 |
high |
high |
2 log |
Table: Comparison of different labelling methods