Proteins undergo many kinds of post translational modifications (PTMs), such as phosphorylation, ubiquitination, SUMOylation, geranyl-geranylation, farnesylation, myristoylation, acetylation, succinylation and palmitoylation. PTMs can be reversible or irreversible, they can be dynamic or stable. PTMs enable fine tuning of protein function in response to various internal and external signals and therefore they have profound effects on cellular physiology, metabolism, survival and growth.
Recent advances in chemical biology methods and advent of mass spectroscopy to characterize PTMs have enabled global characterization of PTMs for many cell types and organisms. In recent years, there has been an unprecedented increase in our understanding about the role of many PTMs that were previously thought to be insignificant. S-palmitoylation is one such PTM, very little was known about the role of palmitoylation in biology of cells until very recently. Earlier, palmitoylation of proteins was studied by labelling cells with radiolabeled fatty acids, such as tritiated or Iodinated fatty acids. The methods were not only cumbersome, but they required expertise to handle radioactive material and suffered from issues like long exposure times, low sensitivity and low throughput.
In recent years, application of advanced techniques and mass spectroscopy to study protein palmitoylation has ushered a new era in the area of investigation of its role in biology of cells. Identification of palmitoylated proteins at global scale has helped molecular biologists, biochemists and protein chemists to understand the scope and biological relevance of this post translational modification. Moreover, the literature about protein palmitoylation is growing every year. This sudden outburst in information about the role of protein palmitoylation has been possible only due to newer methods to capture palmitoylated proteins from cells and tissues.
Why study Palmitoylation?
Protein palmitoylation also termed as S-acylation refers to modification of thiols of cysteine residues by saturated long chain fatty acids such as palmitic acid. The palmitoylation affects biochemical properties of proteins in several ways and therefore, the effect of palmitoylation is pleiotropic. Palmitoylation is catalysed by a family of well conserved enzymes known as DHHC palmitoyl transferases or DHHC-PATs. DHHC-PATs catalyse thio-esterification of cysteine thiols by palmitic acid using palmitoyl coenzyme A as substrate. The reverse process, depalmitoylation is carried out by thioesterases or depalmitoylases, they bring about breaking of thioester bond to release palmitic acid from the protein. The concerted activities of these enzymes in vivo determine the steady state of palmitoylated proteins. The half-life of palmitoylated state can range from few seconds to many hours depending upon the activities of the two enzymes.
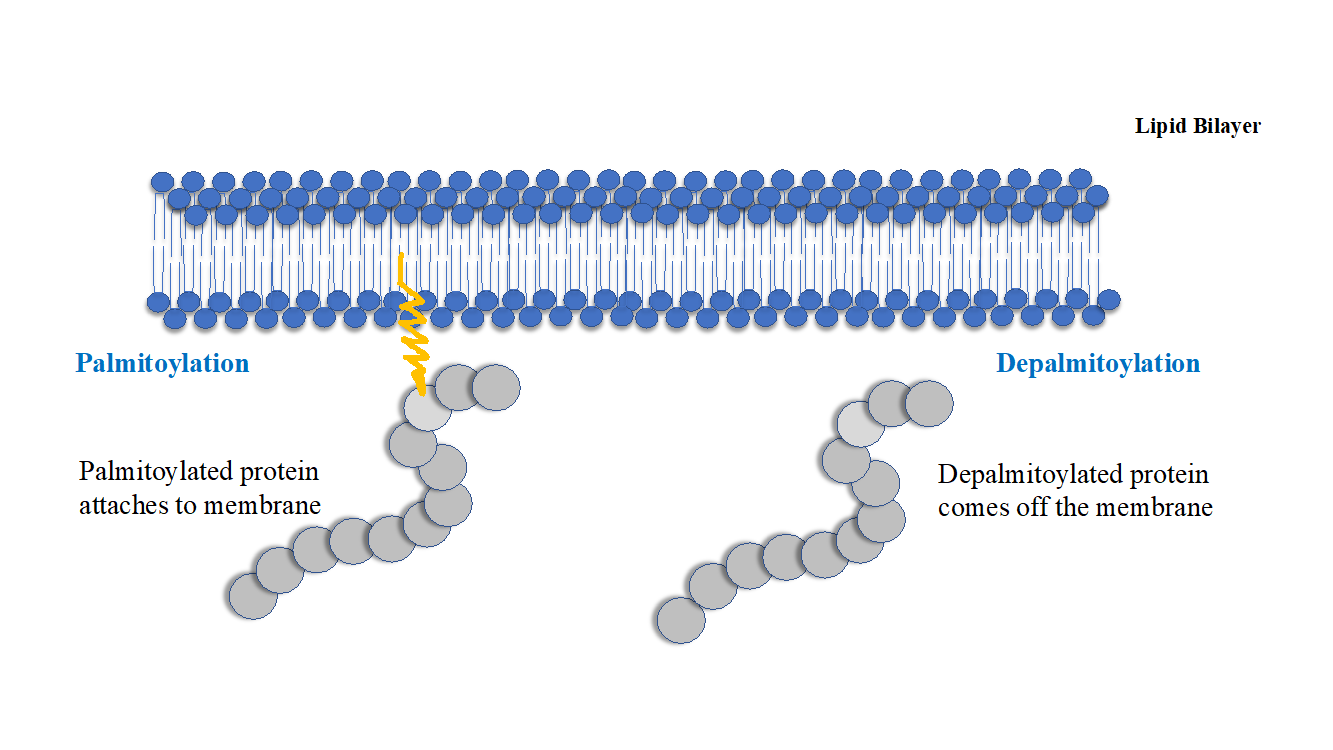
Figure 1: Palmitoylation of soluble cytosolic proteins make them hydrophobic so that they attach/localize to the membranes. The reverse process depalmitoylation brings about removal of palmitic acid residue from the proteins and their return to cytosol. Palmitoylation can therefore, can act as a switch to transiently alter the localisation of water-soluble proteins between cytosol and membranes.
Palmitoylation confers hydrophobic character to the soluble cytosolic proteins, many other PTMs such as farnesylation, geranyl geranylation and myristoylation can also confer hydrophobic character to the soluble proteins but palmitoylation has two attributes which make it different from these acyl/lipid modifications, 1. Palmitoylation is reversible, 2. Hydrophobic character conferred by palmitoylation is almost 5 times more efficient than farnesylation and 100 times more efficient than geranyl-geranylation in retaining proteins at membranes.
Palmitoylation is also a determinant for subcellular localisation of proteins, a well-studied example is Ras GTPases, Ras are GTP hydrolyzing molecules that are present on plasma membrane and initiate downstream signaling in response to external signals affecting cell surface receptors. Ras have three isoforms, H-Ras, N-Ras and K-Ras, these isoforms are indistinguishable in in-vitro assays but have very distinct biochemical behavior in-vivo, this is primarily because of their distinct subcellular localizations in the cells. Ras isoforms differ in their C terminal Hypervariable regions, which contains motif and sequences for lipidation. Lipidation regulates overlapping but distinct localisation of Ras isoforms at plasma membranes and organelles, which in turn influence their interacting partners and varying local concentrations of effectors and activators in the cell. All Ras isoforms are farnesylated in their hypervariable C-terminus region but farnesylation alone does not provide enough hydrophobic glue to remain attached to membrane, Ras isoforms require palmitoylation to achieve stable membrane localisation and to perform shuttling between Golgi complex and plasma membrane.
For many proteins palmitoylation has been found to regulate in-vivo half-life or stability against proteasomal degradation. Palmitoylation has been found to counter ubiquitin mediated proteasomal degradation of proteins. This effect can have serious implications, palmitoylated proteins, which includes transcription factors, kinases, phosphatases, cyclins or other signaling molecules, continue to remain active for longer and their intracellular abundance is likely to be enhanced by palmitoylation. Palmitoylation has been found to be involved in cancer and regulation of cell polarity (Scribble, Ras etc), Regulation of signaling from membranes, which is relevant in events like T cell activation (Lyn and Fck kinases undergo palmitoylation), regulation of calcium signaling (Ryanodine receptors, a calcium exit point from ER), GPCR signaling (Ga subunits are palmitoylated), Neurodegeneration (Huntington protein requires palmitoylation for correct function), vision (visual pigment rhodopsin undergoes palmitoylation). The list can continue to go on but the key point here is that palmitoylation is much more pervasive and plays an important role in biology of cells. We have started accumulating information about palmitoylation recently, but this is nothing more than mere scratching of the surface.
The new methodologies have enabled identification of novel palmitoylated proteins and in this current article we try to describe them in a simplified way.
Acyl Biotinyl Exchange or ABE
The original method was first published by Drisdel and Green in 2004, this was the first non-radioactive approach to study protein palmitoylation and exploited sulfhydryl chemistry or chemistry of thiols to detect protein palmitoylation in a quantitative manner. The method was later on developed and championed by Nicholas Davis’ lab who made improvements to the existing protocol of Drisdel and Green. They also employed mass-spectroscopy in tandem with the method to identify all the palmitoylated proteins in the proteome.
Acyl Biotinyl exchange (ABE) used hydroxylamine base to cleave thioester bond between the sulfhydryl group of palmitoylated cysteine and palmitic acid and uses sulfhydryl reactive biotinylation agents to append an affinity handle to the site of palmitoylation to enable capturing of protein using streptavidin-biotin affinity. In essence the protocol exchanges acyl or palmitic acid moiety with biotin and hence the name acyl biotinyl exchange. Following are key steps of ABE.
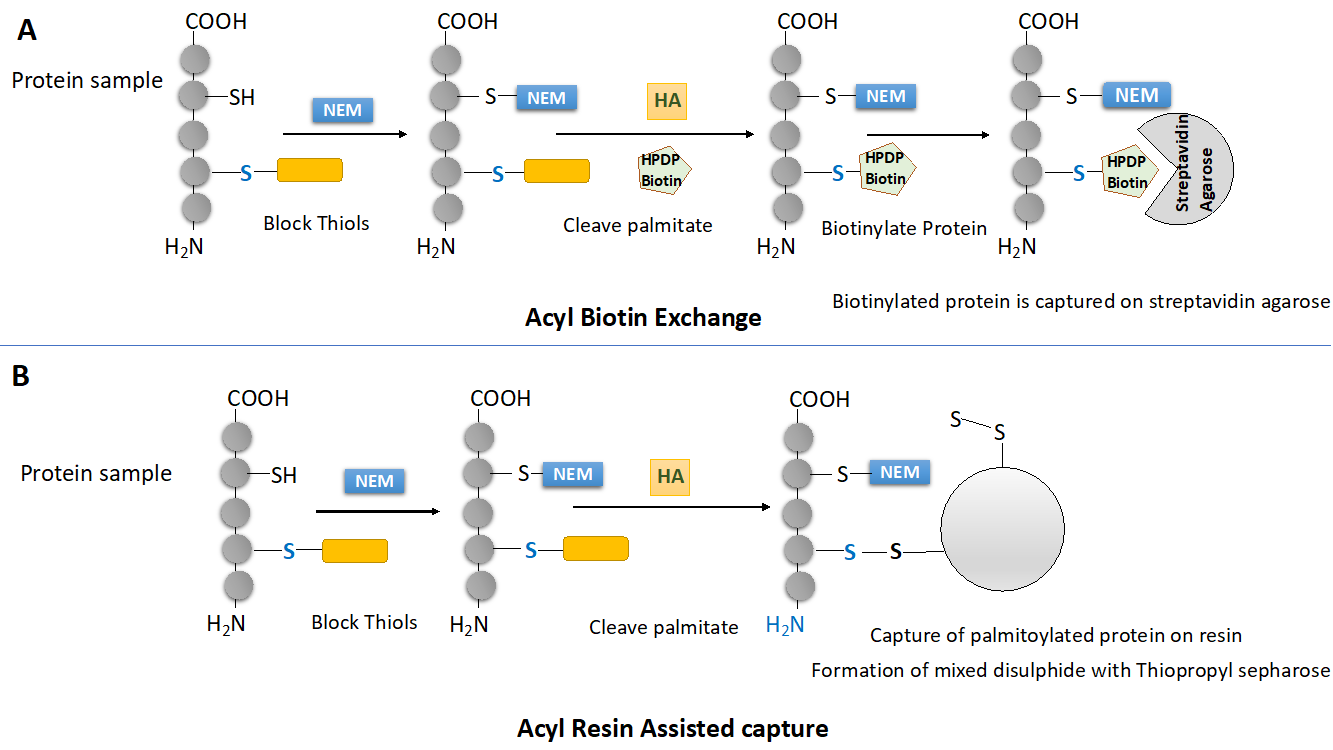
Figure 2: Contemporary methods to capture palmitoylated proteins (A) Outline of Acyl biotinyl Exchange or ABE protocol (B) Outline of Acyl Resin Assisted Capture or Acyl-RAC protocol.
-
Chemical blocking of free thiols
Not all cysteine residues of a protein are palmitoylated, In fact, the cysteines that undergo palmitoylation on a protein are very well conserved. Before cleaving the palmitoyl groups attached to palmitoylated cysteine using hydroxylamine, it is necessary to block free thiols on proteins because we are going to use thiol reactive biotinylation reagents later in the protocol (See Figure 2A). To block free thiols, thiol reactive agents such as NEM (N-ethylmaleImide) or MMTS (Methyl methanethiosulfonate) are used. The chemical blocking of thiols is done in presence of SDS (2%) and Triton X-100 (1%). The SDS denatures the proteins and straightens protein molecules into straight rods, Triton X-100 ensure solubility of hydrophobic palmitoylated proteins.
-
Hydroxylamine assisted cleavage of thioester bonds
Hydroxylamine (neutral pH) cleaves thioester bonds between palmitoyl moiety and sulfhydryl groups. The cleavage of thioester bond generates nascent thiols which are highy reactive.
-
Biotinylation of nascent thiols
Nascent thiols are highly reactive and therefore step 2 is concomitant with step 3. Thiol reactive biotinylating agents such as Biotin BMCC (1-biotinamido-4-[4′- (maleimidomethyl) cyclohexanecarboxamido] butane) or Biotin HPDP (N-[6-(Biotinamido)hexyl]-3¢-(2¢-pyridyldithio) propionamide) are used. Biotin HPDP is preferred because of its higher selectivity. The nascent thiols gets biotinylated.
-
Capture of biotinylated proteins on streptavidin agarose.
Once the proteins have been biotinylated they can be captured using streptavidin agarose beads. Since proteins are biotinylated at cysteines that were previously palmitoylated, tryptic cleavage of biotinylated proteins followed by mass spectroscopic analysis can lead to identification of residues that undergo palmitoylation in a given protein. The streptavidin biotin interaction is too strong to elute palmitoylated proteins from the column and therefore the disulphide bond that forms between Biotin HPDP and thiol is reduced by DTT or 2-mercaptoethanol to finally elute proteins from the streptavidin agarose column.
Acyl Resin assisted capture or Acyl RAC
Another methodology gaining acceptance as a standard method to capture palmitoylated proteins is acyl resin assisted capture or Acyl RAC. This technique successfully replicates results generated using ABE. This technique depends on covalent chromatography and exploits thiol or sulfhydryl chemistry of proteins like ABE but differs in some aspects. This method uses a specific resin, thiopropyl sepharose which can form mixed disulphide with proteins via available thiols (Figure 2B)
The steps common between ABE and Acyl RAC are (1), chemical blocking of free thiols (2), Hydroxylamine assisted cleavage of palmitic acid from the cysteine residues. However Acyl RAC do not require biotinylation of cysteine residues since the nascent thiols generated react directly to the thio-propyl sepharose forming mixed disulphides. The disulphide bonds of mixed disulphides can be reduced using DTT or 2-b-ME (Figure 2B).
Merits and Demerits of the two protocols.
The common merits of both the protocols are, ability to capture palmitoylated proteins by non-radioactive means, quantitative analysis of palmitoylation and high through-put nature of the two protocols. The common demerit of both the protocol is that their success depends on successful blocking of free thiols. Any failure to do so will result in false positives, since both the protocols use thiol-chemistry to capture palmitoylated proteins. Since 100% blocking of free thiols is not possible, false positives is a problem with both the protocols.
ABE requires multiple precipitation and solubilisation steps, which are done to ensure removal of NEM before hydroxylamine cleavage and to remove unreacted biotin HPDP before enrichment on streptavidin agarose, which leads to loss of proteins at each step. Therefore low abundance proteins are lost from palmitomes identified on basis of ABE. Acyl RAC doesn’t require as many precipitation steps and reproduces data generated by ABE with high fidelity and therefore Acyl RAC is gradually replacing ABE.